Microscopia a foglio di luce
La microscopia a foglio di luce, in inglese Light Sheet Fluorescence Microscopy (LSFM) o Selective Plane Illumination Microscopy (SPIM), è una tecnica di microscopia a fluorescenza presentata nel 2004 da Jan Huisken in cui i rami di illuminazione e di raccolta di segnale dell'apparato di misura sono ortogonali l'una all'altra.[1] L'illuminazione è tale per cui la sorgente laser venga focalizzata solo su di un piano del campione, ottenendo così sezionamento ottico, il che può essere ottenuto in diversi modi, tra cui i più comuni prevedono l'utilizzo di lente cilindrica[2] o di modulatori spaziali di luce.[3] Per questa ragione, la tecnica presentata è caratteratterizzata da una maggiore velocità di acquisizione rispetto a tecniche di scanning puntuali (come ad esempio in microscopia cofocale) e una minore quantità di energia rilasciata al campione per unità di superficie, rendendola così uno strumento di analisi adatta allo studio di organismi viventi in fase di sviluppo su scale temporali biologicamente lunghe.[4]

Storicamente, questa tecnica fu ideata da Richard Adolf Zsigmondy e Henry Siedentopf nel 1902, per la visualizzazione di nanoparticelle d’oro, utilizzando come sorgente la luce solare. Dal 1994, LSFM si è sviluppata sulla base di questa tecnica, utilizzando sorgenti laser e campioni biologici fluorescenti, prima con il nome di Orthogonal Plane Fluorescence Optical Sectioning (OPFOS) microscopy[5] e poi nel 2004 con il nome di Selective Plane Illumination Microscopy (SPIM). Dal 2004 ad oggi sono state implementate diverse variazioni e miglioramenti, volte ad allargare il campo di vista[6][7] o a velocizzarne l'acquisizione.[8]
Con la LSFM, si può ottenere l’immagine di un intero piano dell’interno del campione quindi, facendo traslare quest'ultimo attraverso il light-sheet oppure spostando il light-sheet stesso, si può sequenzialmente illuminare ogni suo piano, producendo una serie di immagini a diverse profondità. Da questi dati, la ricostruzione dell’organizzazione e dinamica delle proteine o delle strutture di interesse può essere ricostruita tramite sistemi di analisi e di ricostruzione.[9]
Struttura
modifica
Come già anticipato, nei sistemi LSFM l’illuminazione è posta perpendicolarmente alla parte relativa alla raccolta di luce, cosicché il sistema possa complessivamente essere suddiviso in due parti che spazialmente sono sovrapposte solo in corrispondenza della camera contenente il campione.
Illuminazione
modificaApproccio con lente cilindrica
modificaIn LSFM, lo schema di illuminazione più semplice possibile consta semplicemente di 3 elementi: una sorgente laser, un collimatore ed una lente cilindrica. Molto spesso però si ricorre anche a sistemi di relay per espandere o restringere il fascio in modo tale che sia esteso su di una superficie prestabilita, o a obiettivi da microscopio per ottenere un foglietto di luce ancora più stretto. È vero infatti che lenti cilindriche permettono di mantenere i costi contenuti, ma è anche vero che queste non siano corrette per effetti di curvatura di campo ed altre aberrazioni, e non garantiscano un beam waist inferiore a qualche micron a causa della limitata apertura numerica.
In base a quanto detto e alle leggi sulla propagazione dei fasci gaussiani è possibile quantificare le dimensioni che il fascio di illuminazione avrà nel punto di fuoco dell'ottica utilizzata:
,


lo spessore del light-sheet generato infatti determina la capacità di sezionamento ottico del campione e quindi, in prima approssimazione, la risoluzione assiale del sistema. Un obiettivo con apertura numerica maggiore produrrà un light-sheet di spessore minore, sezionando più finemente il campione.
D'altro canto però un beam waist piccolo (cioè un foglietto molto stretto) determina un parametro di Rayleigh più corto, e quindi una superficie di imaging inferiore. Questo parametro infatti quantifica quanto il fascio gaussiano sia considerabile un foglio di larghezza limitata. In particolare tale parametro vale , ottenuto ponendo il profilo del fascio uguale a . Solitamente si vuole che questo parametro si uguale alla metà della sezione del campione che si vuole analizzare.


Approccio con lente tunabile
modificaCon il progressivo rilascio di elementi ottici sempre più sofisticati e versatili, sono state ideate nuove modalità di scansione a foglio di luce.
Una di queste prevede l'utilizzo di un elemento ottico tunabile, in particolare di una lente, in grado di cambiare potere ottico al variare di una corrente elettrica che la comanda. Al variare della focale della lente tunabile infatti il punto di spessore minimo del foglietto viene traslato avanti e indietro sul piano del campione.[1] Se questo movimento (di solito sulla scala delle centinaia di microsecondi) è fatto su dinamiche temporali più brevi rispetto all'acquisizione del singolo piano da parte del sensore (dell'ordine delle decine di millisecondi), il risultato sarà l'acquisizione del segnale eccitato su di un singolo piano, come quello dato da una lente cilindrica.
Tale approccio ovviamente è più costoso rispetto all'impiego della sola lente cilindrica, richiede inoltre un perfetto sincronismo tra illuminazione e detection, per esempio attraverso l'ultizzo di microcontrollori; ma permette di ottenere un foglietto di dimensioni maggiori rispetto al caso precedente, aumentando così l'area del campione analizzata.
Approccio con specchio galvanometrico
modificaSpecchio galvanometrico per la generazione del foglietto di luce
modifica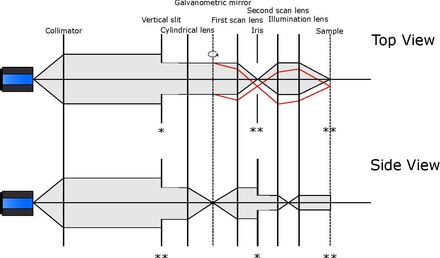
Come nel caso precedente, l'impiego di altri elementi può permettere l'illuminazione di un singolo piano del campione. In particolare, si può accoppiare uno specchio galvanometrico con ad esempio un obiettivo ottico. Il primo è uno specchio, generalmente metallico, in grado di ruotare molto velocemente attorno a uno o più assi. Traslando il fascio laser in entrata, è possibile quindi spostare il punto di fuoco del fascio stesso dato dal solo obiettivo di illuminazione (senza quindi la presenza di un elemento cilindrico), ottenendo un risultato analogo a quanto riportato sopra. Ancora una volta, costi maggiori e difficoltà implementative sono giustificati da una maggiore velocità di scansione, che raggiunge picchi di centinaia di mm/s[10], e da una correzione per eventuali aberrazioni introdotte dal movimento del fascio.
Specchio galvanometrico per il movimento del foglietto di luce
modifica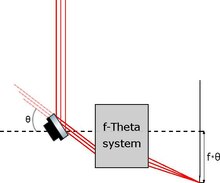
Esistono invece microscopi a foglio di luce disponibili commercialmente in cui lo schema di illuminazione prevede comunque l'uso di uno specchio galvanometrico, ma per traslare un foglietto già generato da una lente cilindrica e non per generare lo stesso come nel paragrafo precedente.
Il movimento del foglietto è possibile grazie ad una particolare configurazione schematica chiamata sistemaf-theta, in cui un tilt angolare di uno specchio è direttamente proporzionale (con costante di proporzionalità pari alla focale del sistema) allo spostamento lineare del punto di fuoco.
Questo schema di illuminazione è particolarmente usato per lo studio dell'interazione di un particolare tipo di campione biologico con qualche stimolo esterno.[2] Spostando infatti il piano illuminato, è possibile ottenere una ricostruzione volumetrica dell'oggetto dell'analisi senza muovere lo stesso e senza quindi indurre shock meccanici e ulteriori stimoli. Ovviamente però spostando il piano illuminato, occorre mantenere quest'ultimo a fuoco sul sensore, il che può essere ottenuto in due modi:
- utilizzo di una lente elettrotunabile in raccolta,[8]
- utilizzo di un traslatore col quale muovere l'obiettivo di raccolta, di modo tale che la distanza tra il piano di entrata dell'obiettivo e il piano illuminato sia sempre pari alla sua distanza di lavoro.
Ancora una volta, l'impiego di più elementi ne complica il controllo e il sincronismo temporale tra questi.
Approcci ibridi
modificaGli schemi di illuminazione proposti fino ad ora seguono lo schema generale di illuminazione di un microscopio a foglio di luce, che prevede, a patto di cambiarne alcuni elementi, l'ortogonalità con il ramo di raccolta. In ogni caso esistono approcci che modificano questo schema, introducendo altri rami di illuminazione[11] o modificando l'angolo tra eccitazione e detection.[12]
Questi approcci sono volti all'aumentare il rapporto segnale rumore delle immagini acquisite e alla riduzione degli effetti di danneggiamento ottico indotti.

Raccolta
modificaAl di là della sua posizione rispetto al ramo di illuminazione, la raccolta del segnale di fluorescenza segue lo schema di un microscopio standard di fluorescenza.
Questa è composta da un obiettivo ottico per la raccolta dei fotoni dal campione, da una lente di tubo, da un filtro di fluorescenza (passa basso o passa banda a seconda del fluoroforo presente nel campione) e da un sensore.
Ognuno degli elementi citati è da valutare a seconda di ciò che si vuole esaminare. In particolare, parametri come magnificazione, risoluzione laterale, quantità di luce raccolta e campo di vista, sono determinati dall'obiettivo usato; qualità del campionamento e velocità di acquisizione dalla camera; reiezione alla sorgente dalla banda del filtro usato.
In particolare la risoluzione laterale del sistema sarà data dalla formula di Abbe:
,

dove al numeratore si ha la lunghezza d'onda emessa dal campione, al denominatore l'apertura numerica dell'obiettivo impiegato.
Per valutare invece la risoluzione assiale del sistema, non è sufficiente valutare quella data dall'obiettivo stesso, ma occorre tenere in conto anche il campionamento geometrico dato dalla matrice di pixel del sensore, ed anche lo spessore del foglietto di luce.
Considerando un sistema formato da camera, obiettivo e lente di tubo, la sua profondità di campo è:

dove DOF sta per depth of field, n per indice di rifrazione del mezzo in cui è contenuto il campione, M per magnificazione ed e per la dimensione del pixel.
La risoluzione assiale del sistema con illuminazione a folgio di luce sarà dato dalla convoluzione di questa con il profilo di intensità del light-sheet stesso.[13]

Estensione della profondità di campo
modificaSoprattutto quando si utilizzano obiettivi ad alta magnificazione e apertura numerica, la profondità di campo può non essere abbastanza estesa per ottenere un'immagine volumetrica del campione senza muovere lo stesso. Per questo motivo sono state ideate tecniche per estedere la profondità di campo, senza intaccare la risoluzione del risultato finale rispetto al caso non esteso.
La prima tecnica prevede l'impiego di elementi a proprietà ottiche variabili come una lente elettrotunabile in un particolare punto del ramo di raccolta.[8]
Altre tecniche invece prevendono l'introduzione controllata nel sistema di aberrazioni ottiche, così da poter, una volta acquisite le immagini del campione, deconvolverle con la point spread function aberrata del sistema, ricostruendo il volume in esame senza effetti indesiderati.[14]
In conclusione, un'ulteriore modalità di estensione della profondità di campo prevede l'inserimento di uno strato di materiale (liquido o solido) diverso rispetto al liquido di imaging per caratteristiche ottiche (in particolare per indice di rifrazione), per sfruttare la Rifrazione del segnale di fluorescenza e riuscire a raccogliere luce da un cono più elungato.[15]
Caratteristiche della LSFM
modificaMontaggio del campione
modifica
La particolare disposizione dei rami di illuminazione e detection in un microscopio a foglio di luce fa sì che il campione debba essere posizionato in maniera diversa rispetto ad altre tecniche di microscopia ottica. Solitamente, un microscopio LSFM è costruito in modo tale che il foglio di luce sia perpendicolare al tavolo ottico, motivo per cui il campione necessita di un montaggio in verticale, come in figura. Ciò non toglie che per la grande varietà di campioni possibili e per le loro caratteristiche, ognuno di questi possa avere necessità diverse. I modi di fissaggio più comuni sono i seguenti:
- Fissaggio del campione tramite colla ad un supporto, in questa modalità è importante che il collante non sia solubile dalla soluzione di immersione del campione stesso,
- Campioni più grandi (come ad esempio il pesce zebra), per attività di misura in vivo, vengono prima sedati e poi posti in una matrice gel che permetta loro la respirazione ma li mantenga fermi. Per problemi di focusing dati da un eventuale tubo in PVC o vetro, è preferibile estrudere la parte di gel col campione, piuttosto che procedere con a misura dentro il tubo,
- Colture di cellule possono essere montate su di un vetrino standard da microscopio, poi da appendere nella cameretta del campione,
- Altri campioni come Arabidopsis thaliana, vengono posti in gel come agarosio o gelrite. Successivamente il gel viene tagliato, per evitare che degradi la qualità dell'imaging a causa di scatterig e assorbimento, sia per quanto riguarda illuminazione che detection.[16]
- Campioni liquidi possono essere montati in piccole sacche di plastica, prestando attenzione che questa abbia indice di rifrazione quasi uguale a quello del liquido usato per il corretto funzionamento dell'obiettivo, onde evitare effetto lente dato da un gradiente d'indice.[17]
Sono stati sviluppati invece alcuni LSFM che sfruttato schemi in cui il foglietto di luce è parallelo al tavolo ottico. Questi consentono l'utilizzo di tecniche di montaggio standard.[18][19][20][21]
Artefatti a strisce
modifica
Siccome la luce penetra nel campione da un lato, eventuali strutture (come ad esempio radici o pigmenti sulla parte esterna), possono degradare il foglietto per assorbimento o scattering. In particolare, l'assorbimento dato da zone con un aumento significativo dell'indice di rifrazione produce zone d'ombra allungate dalla parte opposta rispetto all'illuminazione. Questo fenomeno è una caratteristica distintiva della light sheet fluorescence microscopy e prende il nome di "stripe artefacts"
Esistono fondalmente tre modalità per superare il problema. La prima prevede di cambiare gradualmente l'angolo di incidenza del foglietto, secondo la tecnica che prende il nome di "pivoting". La direzione cambia in maniera repentina (~1 kHz rate), su pochi gradi (~10°), così che si illuminino regioni altrimenti nascoste.[3]
Nella seconda modalità, invece di avere un unico ramo di illuminazione, si procede con l'eccitare segnale nel campione da ambo i lati, così da eliminare l'effeto a strisce d'ombra. Ovviamente questo approccio raddoppia la dose di luce sul campione, aumentandone quindi il danneggiamento.[11]
Sono stati anche implementate routine in cui i due metodi sopra vengono combinati.[22]
In ultimo, esiste un algoritmo chiamato VSNR (Variational Stationary Noise Remover), sviluppato in Fiji, che aiuta a rimuovere tali artefatti.[23]
Note
modifica- ^ a b J. Huisken, Optical Sectioning Deep Inside Live Embryos by Selective Plane Illumination Microscopy, in Science, vol. 305, n. 5686, 13 agosto 2004, pp. 1007-1009, DOI:10.1126/science.1100035. URL consultato il 1º luglio 2019.
- ^ a b Alex Costa, Alessia Candeo e Luca Fieramonti, Calcium Dynamics in Root Cells of Arabidopsis thaliana Visualized with Selective Plane Illumination Microscopy, in PLoS ONE, vol. 8, n. 10, 16 ottobre 2013, pp. e75646, DOI:10.1371/journal.pone.0075646. URL consultato il 1º luglio 2019.
- ^ a b Chiara Garbellotto e Jonathan M. Taylor, Multi-purpose SLM-light-sheet microscope, in Biomedical Optics Express, vol. 9, n. 11, 12 ottobre 2018, p. 5419, DOI:10.1364/boe.9.005419. URL consultato il 1º luglio 2019.
- ^ (EN) Michael W. Adams, Andrew F. Loftus e Sarah E. Dunn, Current Protocols in Cytometry, John Wiley & Sons, Inc., 5 gennaio 2015, pp. 12.37.1–12.37.15, DOI:10.1002/0471142956.cy1237s71, ISBN 978-0-471-14295-9. URL consultato il 1º luglio 2019.
- ^ Jan A. N. Buytaert, Emilie Descamps e Dominique Adriaens, The OPFOS Microscopy Family: High-Resolution Optical Sectioning of Biomedical Specimens, in Anatomy Research International, vol. 2012, 3 novembre 2012, pp. 1-9, DOI:10.1155/2012/206238. URL consultato il 1º luglio 2019.
- ^ Rory M. Power e Jan Huisken, Adaptable, illumination patterning light sheet microscopy, in Scientific Reports, vol. 8, n. 1, 25 giugno 2018, DOI:10.1038/s41598-018-28036-2. URL consultato il 1º luglio 2019.
- ^ Sébastien Wolf, Willy Supatto e Georges Debrégeas, Whole-brain functional imaging with two-photon light-sheet microscopy, in Nature Methods, vol. 12, n. 5, 29 aprile 2015, pp. 379-380, DOI:10.1038/nmeth.3371. URL consultato il 2 luglio 2019.
- ^ a b c Florian O. Fahrbach, Fabian F. Voigt e Benjamin Schmid, Rapid 3D light-sheet microscopy with a tunable lens, in Optics Express, vol. 21, n. 18, 30 agosto 2013, p. 21010, DOI:10.1364/oe.21.021010. URL consultato il 1º luglio 2019.
- ^ (EN) Johannes Schindelin, Ignacio Arganda-Carreras e Erwin Frise, Fiji: an open-source platform for biological-image analysis, in Nature Methods, vol. 9, n. 7, 2012-7, pp. 676-682, DOI:10.1038/nmeth.2019. URL consultato il 1º luglio 2019.
- ^ Giuseppe Sancataldo, Vladislav Gavryusev e Giuseppe de Vito, Flexible Multi-Beam Light-Sheet Fluorescence Microscope for Live Imaging Without Striping Artifacts, in Frontiers in Neuroanatomy, vol. 13, 8 febbraio 2019, DOI:10.3389/fnana.2019.00007. URL consultato il 2 luglio 2019.
- ^ a b Jan Huisken e Didier Y. R. Stainier, Even fluorescence excitation by multidirectional selective plane illumination microscopy (mSPIM), in Optics Letters, vol. 32, n. 17, 27 agosto 2007, p. 2608, DOI:10.1364/ol.32.002608. URL consultato il 2 luglio 2019.
- ^ Berl R Oakley, Faculty of 1000 evaluation for LITE microscopy: Tilted light-sheet excitation of model organisms offers high resolution and low photobleaching., su F1000 - Post-publication peer review of the biomedical literature, 7 maggio 2018. URL consultato il 2 luglio 2019.
- ^ MERTZ, JEROME., INTRODUCTION TO OPTICAL MICROSCOPY., CAMBRIDGE UNIV PRESS, 2019, ISBN 1-108-42830-4, OCLC 1084422001. URL consultato il 3 luglio 2019.
- ^ Pantazis Mouroulis, Depth of field extension with spherical optics, in Optics Express, vol. 16, n. 17, 11 agosto 2008, p. 12995, DOI:10.1364/oe.16.012995. URL consultato il 4 luglio 2019.
- ^ Raju Tomer, Matthew Lovett-Barron e Isaac Kauvar, SPED Light Sheet Microscopy: Fast Mapping of Biological System Structure and Function, in Cell, vol. 163, n. 7, 2015-12, pp. 1796-1806, DOI:10.1016/j.cell.2015.11.061. URL consultato il 3 luglio 2019.
- ^ Alexis Maizel, Daniel von Wangenheim, Fern n Federici, Jim Haseloff, Ernst H.K. Stelzer, High-resolution live imaging of plant growth in near physiological bright conditions using light sheet fluorescence microscopy, in The Plant Journal, vol. 68, n. 2, October 2011, pp. 377-385, DOI:10.1111/j.1365-313X.2011.04692.x, ISSN 0960-7412, PMID 21711399.
- ^ T. Wohland, X. Shi, J. Sankaran e E. H. Stelzer, Single plane illumination fluorescence correlation spectroscopy (SPIM-FCS) probes inhomogeneous three-dimensional environments., in Optics Express, vol. 18, n. 10, May 2010, pp. 10627-10641, Bibcode:2010OExpr..1810627W, DOI:10.1364/oe.18.010627, PMID 20588915.
- ^ J. Capoulade, M. Wachsmuth, L. Hufnagel e M. Knop, Quantitative fluorescence imaging of protein diffusion and interaction in living cells, in Nature Biotechnology, vol. 29, n. 9, 2011, pp. 835-839, DOI:10.1038/nbt.1928, PMID 21822256.
- ^ Terrence F. Holekamp, Diwakar Turaga e Timothy E. Holy, Fast Three-Dimensional Fluorescence Imaging of Activity in Neural Populations by Objective-Coupled Planar Illumination Microscopy, in Neuron, vol. 57, n. 5, 13 marzo 2008, pp. 661-672, DOI:10.1016/j.neuron.2008.01.011, ISSN 0896-6273, PMID 18341987.
- ^ Y. Wu, A. Ghitani, R. Christensen, A. Santella, Z. Du, G. Rondeau, Z. Bao, D. Colon-Ramos e H. Shroff, Inverted selective plane illumination microscopy (iSPIM) enables coupled cell identity lineaging and neurodevelopmental imaging in Caenorhabditis elegans, in Proceedings of the National Academy of Sciences, vol. 108, n. 43, 25 ottobre 2011, pp. 17708-17713, Bibcode:2011PNAS..10817708W, DOI:10.1073/pnas.1108494108, ISSN 0027-8424, PMC 3203761, PMID 22006307.
- ^ Yicong Wu, Peter Wawrzusin, Justin Senseney, Robert S Fischer, Ryan Christensen, Anthony Santella, Andrew G York, Peter W Winter, Clare M Waterman, Zhirong Bao, Daniel A Colón-Ramos, Matthew McAuliffe e Hari Shroff, Spatially isotropic four-dimensional imaging with dual-view plane illumination microscopy, in Nature Biotechnology, vol. 31, n. 11, 2013, pp. 1032-1038, DOI:10.1038/nbt.2713, ISSN 1087-0156, PMC 4105320, PMID 24108093.
- ^ Jan Huisken e Didier Y. R. Stainier, Even fluorescence excitation by multidirectional selective plane illumination microscopy (mSPIM), in Optics Letters, vol. 32, n. 17, 2007, pp. 2608-10, Bibcode:2007OptL...32.2608H, DOI:10.1364/OL.32.002608, ISSN 0146-9592, PMID 17767321. URL consultato il 27 ottobre 2012.
- ^ Jerome Fehrenbach, Pierre Weiss e Corinne Lorenzo, Variational Algorithms to Remove Stationary Noise: Applications to Microscopy Imaging, in IEEE Transactions on Image Processing, vol. 21, n. 10, 2012, pp. 4420-4430, Bibcode:2012ITIP...21.4420F, DOI:10.1109/TIP.2012.2206037, ISSN 1057-7149, PMID 22752131.